Spatial Phylogenetics Workshop
Dr. Joel H. Nitta
@joelnitta@fosstodon.org
Associate Professor @ Chiba University
Research interests: Ecology and evolution of ferns
Photo: J-Y Meyer

Basic inputs for spatial phylogenetics
- A phylogeny
- Spatial occurrence data


… which are linked by taxonomic names (OTUs)
Sources of occurrence data
- Herbaria or museums
- Floras or checklists
- Previous studies
- Your own data

Online sources of occurrence data
- GBIF
- Kew Plants of the World Online (POWO)
- VertNet
etc…
![]()
![]()
![]()
Types of occurrence data
Can take many forms:
- geometric shapes
- points
- checklists
- your own surveys
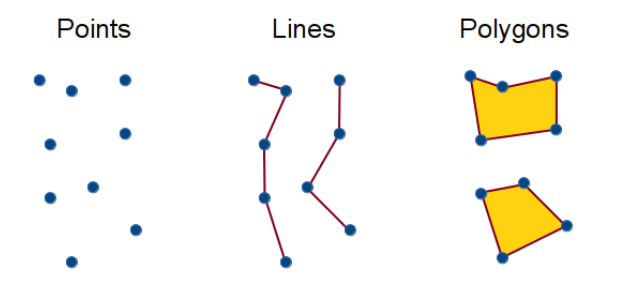
We will focus on point data (the data available from GBIF) during the coding session
GBIF https://www.gbif.org/

Coordinate Reference Systems (CRS)
Geographic Coordinate System (GCS)
- Where the data are located
- Round (like the earth)
- Usually in degrees
Projected Coordinate System (PCS)
- How to draw a map of the data
- Flat (like a piece of paper)
- Usually in meters
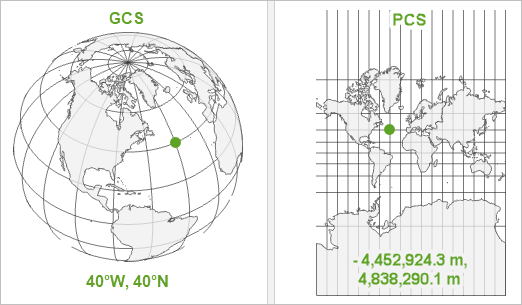
Geographic Coordinate System (GCS)
Latitude and longitude alone are not enough
The earth is not a perfect sphere
GCS defines how to model the earth (e.g., WGS84)

Hiker’s coordinates at 134.577°E, 24.006°S. But where is she (A or B)?
https://www.esri.com/arcgis-blog/products/arcgis-pro/mapping/gcs_vs_pcs/
Projected Coordinate System (PCS)
The earth is round, but we project it onto flat maps
The decision of how to do this is not trivial
There will always be some amount of distortion in area, distance, or direction

https://geoawesomeness.com/wp-content/uploads/2022/03/projections.jpg
Coordinate Reference Systems
You need to choose an appropriate CRS for your study (there are thousands)
If you assume that your sampling units have equal area, make sure to use an equal-area projection (e.g., Mollweide)

The Mollweide projection. The orange dots have the same area, but their shape is distorted as you move away from the equator.
https://en.wikipedia.org/wiki/Mollweide_projection
From occurrences to grid-cells
Raw data are often provided on a per-species basis
But we are interested in assemblages (grid-cells) of species → need to group species together

https://phys.org/news/2018-12-local-conditions.html
Phylogenetic data
- Is there a tree available, or do you need to build it from scratch?

https://images.unsplash.com/
Sources of DNA sequence data
- GenBank
- access in R via the
rentrezpackage- (or
restezpackage for larger datasets)
- (or
https://a-little-book-of-r-for-bioinformatics.readthedocs.io/
Building a tree from scratch
We don’t have time to cover this today - that is a whole topic of study unto itself!

https://mediacdn.nhbs.com/jackets/jackets_resizer_xlarge/17/170234.jpg
Sources of phylogenetic trees
- Previous publications
- R packages that provide trees (
ftolrfor ferns) - Open Tree of Life (
rotlR package) (caution!) - Software that places tips on the tree by taxonomy (caution!)
![]()
https://docs.ropensci.org/rotl/logo.svg
What if I don’t have a tree for my group?
- A tree at the species level may not be necessary. Consider doing the analysis at a higher taxonomic level (e.g., genus)

https://www.irasutoya.com/
Taxonomic issues
- Old names
- Misspelled names
- Mismatching synonyms

We need to resolve names to a standard taxonomic database
Distribution of endemicity
- Paleoendemism
- Refugia
- Colonization by distantly related lineages
- Neoendemism
- Recent speciation
- Mixed endemism
- Multiple processes

Distribution of endemicity
- Paleoendemism
- Refugia
- Colonization by distantly related lineages
- Neoendemism
- Recent speciation
- Mixed endemism
- Multiple processes
Case study: Ferns of Japan
- > 600 species
- Dense sampling
- 10 x 10 km maps of every species
- DNA (rbcL) for > 98% of species
Nitta et al. AJB 2022 https://doi.org/10.1002/ajb2.1848
Photos A. Ebihara

Case study: Ferns of Japan
Variation in climate from N (subarctic) to S (subtropical)
Variation in elevation
Main islands continental, southern islands oceanic
Nitta et al. AJB 2022 https://doi.org/10.1002/ajb2.1848
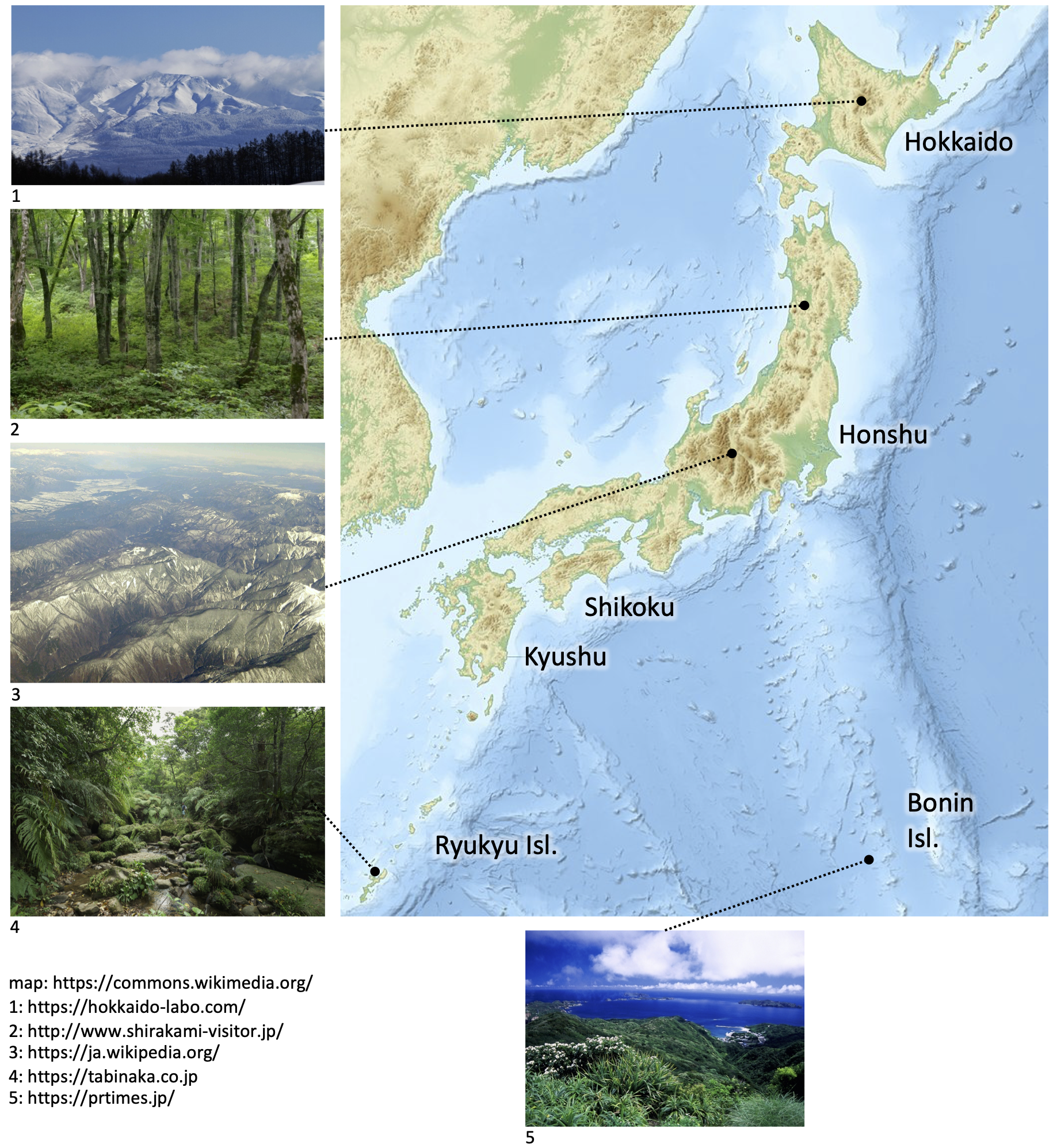
Skewed distribution of endemism
- Southern-most islands are subtropical
- Very different climate from rest of country
- High rates of mixed- and paleo-endemism
- Due to distantly related (tropical) lineages

Reproductive mode as driver of biodiversity
Phylogenetic diversity is predicted by % of apogamous (asexual) species
- Apogamous species tend to be hybrids that share identical plastid sequences with other species

Spatial autocorrelation
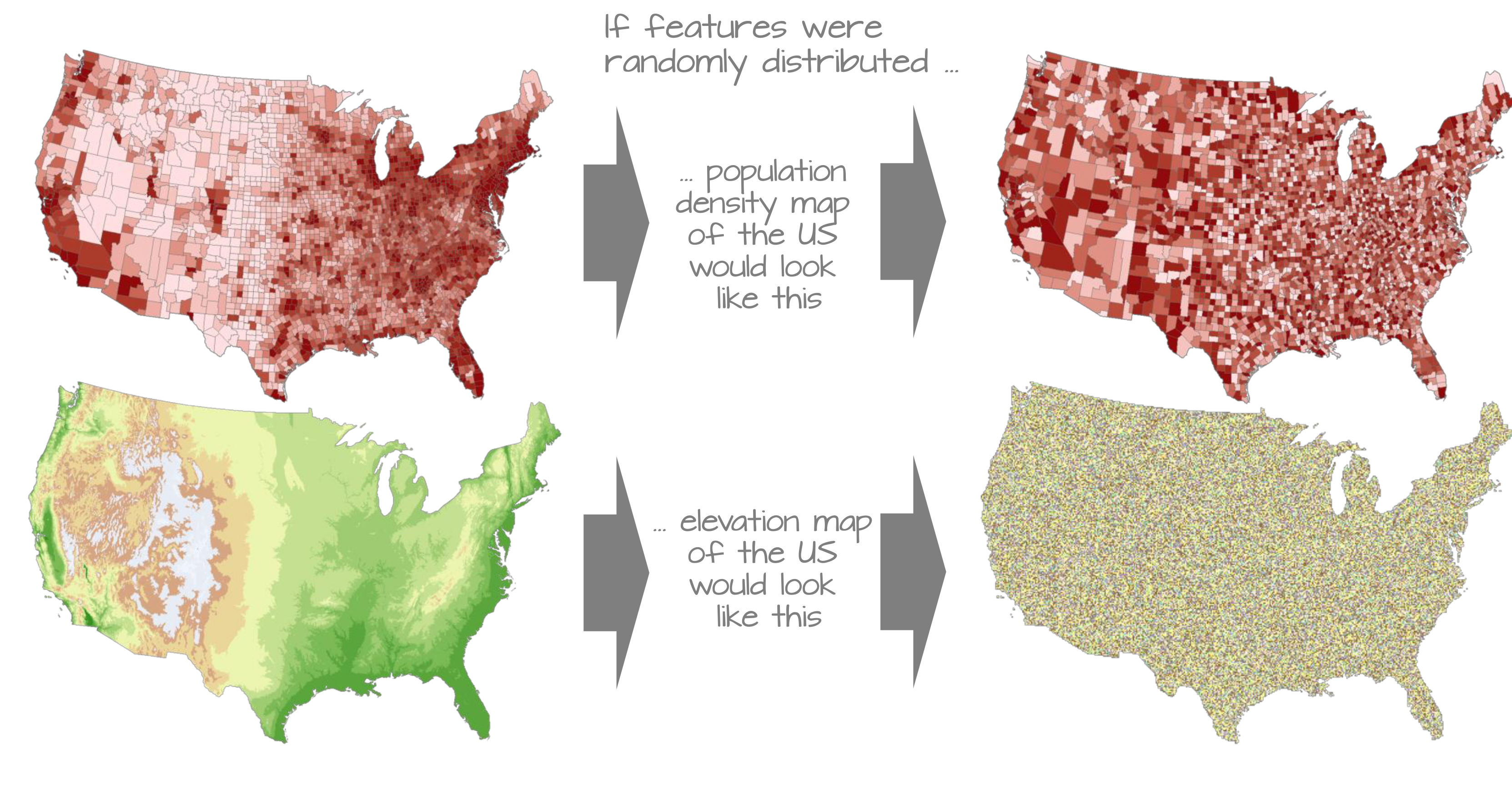
Accounting for spatial autocorrelation
Compare amount of observed autocorrelation to some expected value: Moran’s I

Accounting for spatial autocorrelation
Workflow:
- Conduct non-spatial analysis
- Check degree of Moran’s I in model residuals
- If significant, re-do analysis using spatial model

3-4 regions in ferns of Japan

High rates of endemism on remote islands cause difference in taxonomic and phylogenetic bioregions
Rates of protection vary by biodiversity metric

canaper R package
- Can automate CANAPE analysis with R scripts
- Don’t need to switch between Biodiverse and R (do it all in R)
![]()